
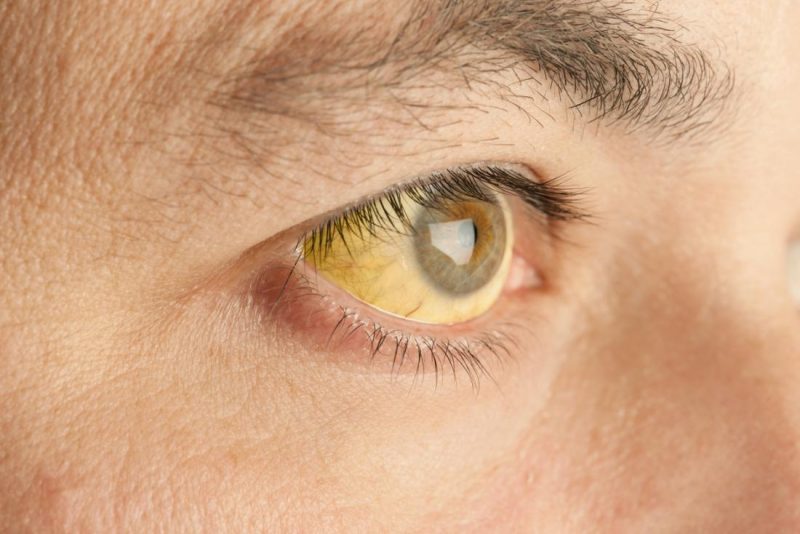
What is Gilbert’s Syndrome?
Gilbert’s syndrome, also known as familial nonhemolytic jaundice or hyperbilirubinemia 1, is a common, benign genetic hepatic dysfunction that occurs when the liver does not process bilirubin correctly (the liver is not able to degrade bilirubin). Therefore, this syndrome is characterized by an excess of bilirubin (a product of red blood cells) in the blood, a condition known as hyperbilirubinemia.
Bilirubin, an orange-yellow pigment, is usually derived from the degradation of the hemoglobin in the dead red blood cells. In normal conditions, the degradation of bilirubin present in the blood is possible only in the liver and under the action of an enzyme called uridine diphosphate glucuronosyltransferase (UGT). This enzyme is able to increase the solubility of bilirubin by the addition one or two molecules of glucuronic acid. After the action of this hepatic enzyme, the bilirubin can be actively transported into the bile and then excretes from the body (in the stool).
Patients with Gilbert’s syndrome have a reduced level of this crucial hepatic enzyme required for elimination of bilirubin, and therefore, they have elevated levels of bilirubin (hyperbilirubinemia). The elevated levels of bilirubin in the blood may produce symptoms such as jaundice (mild yellowing of the mucous membranes, whites of the eyes and skin). Other symptoms that have been associated with Gilbert’s syndrome include abdominal pain, fatigue, and weakness. However, most affected individuals have no symptoms. Importantly, this is an entirely benign condition that is not associated with any increased mortality or any underlying chronic liver disease.
Overall, Gilbert’s syndrome is estimated to occur in 3-7% of the general population in the USA and is much more common among males than females.
Which is the role of genes in the development of Gilbert’s Syndrome?
Gilbert’s syndrome is caused by a modification in the DNA (known as mutation) of the gene known as uridine diphosphate-glucuronosyltransferase-1A1 (UGT1A1). This mutation caused a defective UGT1A1 gene that cannot correctly produce a necessary enzyme described above, called uridine diphosphate glucuronosyltransferase (UGT). Thus, people having Gilbert’s syndrome possess a reduced level of this enzyme required for the elimination of bilirubin, and therefore, they have elevated levels of bilirubin (hyperbilirubinemia).
Usually, this unconjugated bilirubin is taken by the liver and, with the support of the UGT enzyme, is transformed to water-soluble bilirubin (known as conjugated bilirubin), which are then excreted in the bile. When a patient has a mutated gene, its blood contains excess amounts of a specific form of bilirubin (known as unconjugated bilirubin) because the patient does not produce enough quantity of the UGT enzyme, which is essential for the elimination of the bilirubin from the body. Therefore, patients with Gilbert’s syndrome have a deficiency of the UGT enzyme, and therefore, bilirubin is not converted at the standard rate. Thus, the unconjugated form of bilirubin is present at high levels in the bloodstream and can cause jaundice.
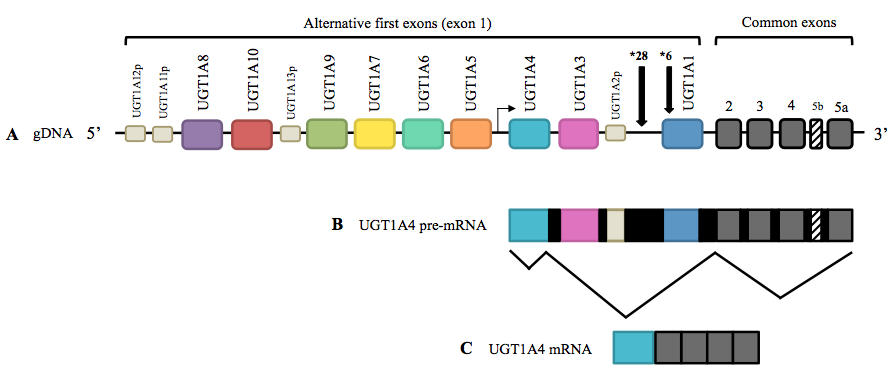
It is important to underline that although the individuals with Gilbert’s syndrome possess around 33% of the regular UGT enzyme activity, they are also able to conjugate enough bilirubin to prevent symptoms from developing. For this reason, patients with Gilbert’s disease are entirely normal with no other symptoms.
The mutated (abnormal) UGT1A1 gene that causes Gilbert’s syndrome is common. Many persons have one copy of this gene in their DNA. However, it is important to underline that two abnormal (mutated) copies of this gene are required to induce Gilbert’s syndrome. This means that DNA mutations in the UGT1A1 gene may be inherited from both parents to develop this syndrome. Therefore, people with only a single abnormal (mutated) gene have higher than normal levels of unconjugated bilirubin but do not have Gilbert’s syndrome.
The most common genetic mutation (permanent alteration in the DNA sequence) that produces Gilbert’s syndrome occurs in an area of DNA near the UGT1A1 gene, known as promoter region, which controls the production of the UGT enzyme. As a result, this genetic mutation impairs the UGT enzyme production.
Importantly, it has also been observed that another type of mutation in the UGT1A1 gene can cause Gilbert’s syndrome (commonly in Asians populations). In this case, the UGT1A1 gene of a patient has a mutation that changes a single protein building block (amino acid) in the UGT enzyme. Also, this type of mutation results in reduced enzyme function.
Additional types of mutations in the UGT1A1 gene can cause a syndrome known as Crigler-Najjar syndrome, which is a more dangerous and severe form of hyperbilirubinemia.
How is Gilbert’s syndrome diagnosed?
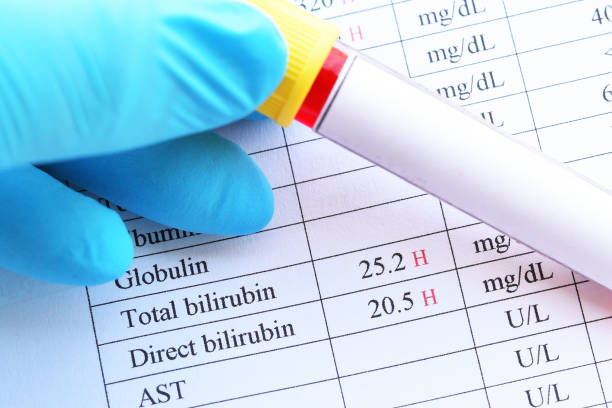
This syndrome can be diagnosed using a blood test to measure the levels of bilirubin in the blood and by performing liver function tests to make sure that there are no other causes of the high bilirubin levels. If the test results show that a person has high levels of bilirubin in the blood, but the liver works normally, a confident diagnosis of Gilbert’s syndrome can usually be made. Sometimes a genetic test could be indispensable to confirm a diagnosis of Gilbert’s syndrome.
Take home messages
Gilbert’s syndrome is an inherited disorder that runs in families (which means that it is hereditary). Patients with this type of illness have a defective UGT1A1 gene which causes the liver to have difficulties eliminating bilirubin from the bloodstream.
Gilbert syndrome is usually mild (not dangerous) and does not cause long-term symptom; therefore, no specific treatments are required for patients with this syndrome. It is a benign liver condition and does not usually cause complications.
However, the patients still have to perform blood test tests and liver function tests in order to prevent the development of some complications such as chronic hepatitis and cholelithiasis.
It is very important to underline that although most affected individuals have no symptoms, the low level of the UGT enzyme that causes Gilbert’s syndrome can also increase the side effects of some drugs (such as for treating cancer or HIV) because these drugs are usually processed by the same bilirubin-processing enzyme (UGT enzyme).

Current position: PostDOC at Technologica Research Institute, Crotone, Italy and Student at School of Specialization in Medical Genetics, University “Magna Graecia” of Catanzaro, Campus “S. Venuta”, Catanzaro, Italy.
Education: University of Calabria, Italy, Degree in Biological Sciences, 2003; University of Milano-Bicocca, Italy, Master in Bioinformatic, 2006; University “Magna Graecia” of Catanzaro, Campus “S. Venuta”, Catanzaro, Italy, PhD in Molecular Oncology, Experimental Immunology and Development of Innovative Therapies, 2012.
Nonscientific interests: Computers and technology, fanta-scientific movies, reading, sports.
Scientific interests focus on medical genetics, molecular and cellular oncology and stem cell biology. I’m a coauthor of 35 published papers, 1 patent and 3 book chapters.
Most Popular
1
Genomics investing 101
-By Gergana Koleva
2
Genomic altruism in the era of Big Data and fragile privacy
-By Gergana Koleva
3
CAR-T therapy: fighting fire with a flood
-By Kathryn McLaughlin
4
Finding Faces in DNA Traces
-By Charlie Lunan
5
“Woke science” in the genomics era
-By Kathryn McLaughlin